
















Three issues have been identified affecting tools for fusion detection, and thereby affecting workflows delivered by the Biomedical Genomic Analysis 20.0 plugin for fusion detection, which contain these tools. The affected workflows are:
Full details of the software, workflows and tools affected are provided in the “Affected software and tools” section below.
These problems were addressed in Biomedical Genomic Analysis 20.0.1 and Biomedical Genomic Analysis Server Plugin 20.0.1 through a combination of bug fixes and improvements.
Some fusions identified are not being annotated correctly by Annotate Fusions with Known Fusion Information: Some known fusions detected in the data are not being annotated, while some unknown fusions detected may be annotated as known fusions.
For two fusions where both breakpoints are within 12bp of one another, a given read can be counted as supporting both those fusions. This can then lead to the two fusions being assigned the same read count, as shown in Figure 1. Closer inspection of the read-mapping may reveal that one of the fusions has much better support than the other.

Figure 1. Two fusions with identical 5′ breakpoints and 3′ breakpoints within 12 bp of each other are listed here. The read count is reported to be the same for both, but inspection of the read mapping could reveal better support for one of them.
Functionality introduced in the Biomedical Genomic Analysis 20.0 plugin to detect exotic fusions, i.e. fusions where one or both breakpoints are not at an exon boundary (Figure 2), has led to a large number of such fusions being detected for some datasets, a large fraction of which are false positives.
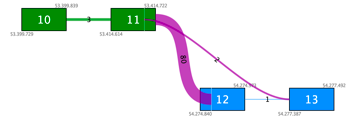
Figure 2. Examples of exotic fusions into the middle of an exon and into an intron
We plan to release an update to the software where these issues will be addressed. In the meantime, possible actions to take when detecting fusions are:
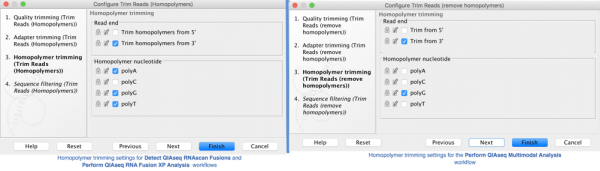
Figure 3. Homopolymer trimming settings included in workflows delivered by Biomedical Genomic Analysis 20.0
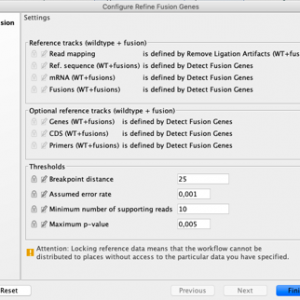
Figure 4. Breakpoint distance parameter of the Refine Fusion Genes tool increased to 25 from the default value of 10.
The issues described on this page were addressed in Biomedical Genomic Analysis 20.0.1 and Biomedical Genomic Analysis Server Plugin 20.0.1 through a combination of bug fixes and improvements.
Affected software:
On affected software versions, the following workflows were affected:
as those workflows contained one or more of the following affected tools:













