As treatments are increasingly tailored to an individual for greater efficacy, understanding the immune repertoire becomes more critical. B-cell receptor (BCR) reconstruction plays a crucial role in understanding the immune system’s response to various stimuli. Our immune system produces B cells, which carry unique receptors on their surfaces. These receptors act like keys, explicitly recognizing and binding to foreign invaders like viruses or bacteria. By reconstructing BCRs from single-cell RNA-seq (scRNA-seq) data, we can gain valuable information on the diversity and specificity of the immune response at the single-cell level.
Have you ever had to put a jigsaw puzzle together? That’s what happens when BCRs are reconstructed from scRNA-seq data. Choosing the right tool to assemble your numerous small pieces of data then becomes vital for achieving accurate results. But with so many software options out there, which one takes the crown?
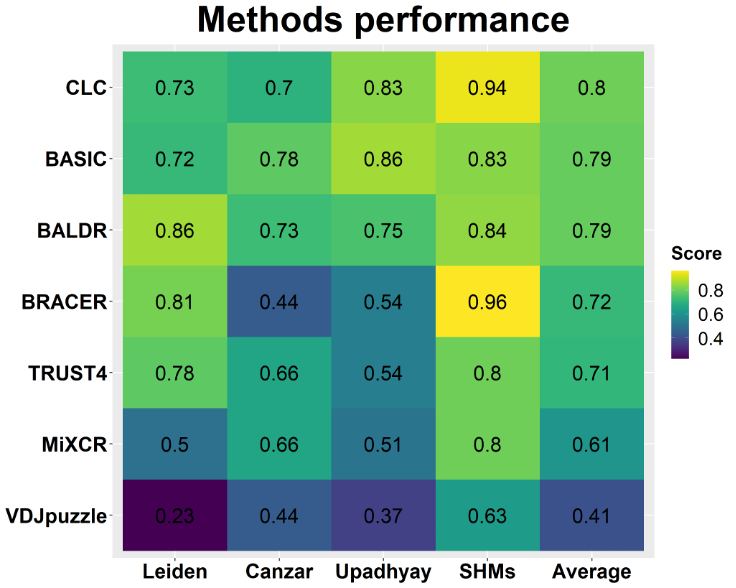
A recent publication (1) compared the performance of several popular tools for BCR reconstruction with scRNA-seq data, namely:
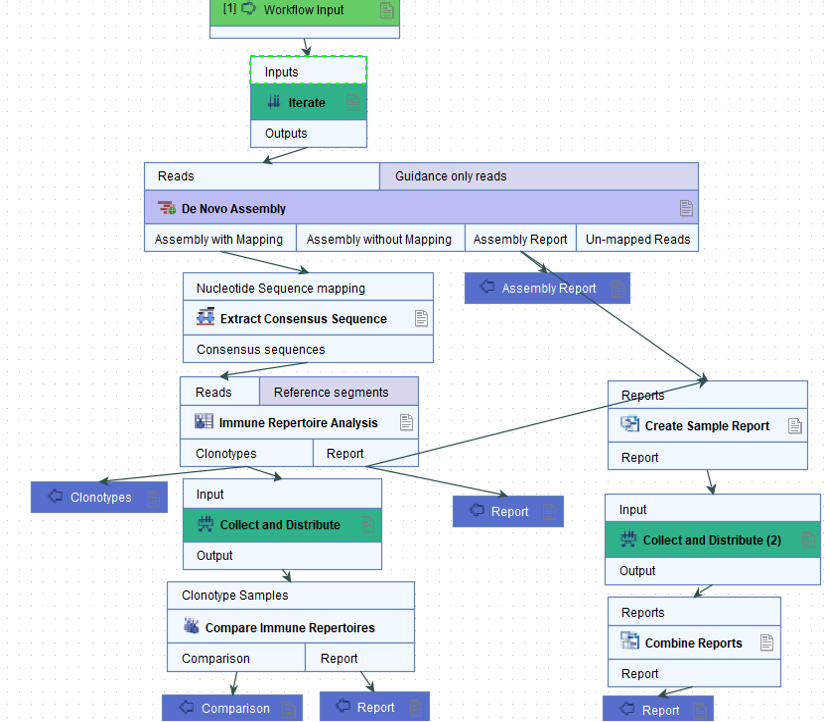
Abdul R. Estalefi and Mathias Østergaard Mikkelsen, students of the Department of Biological and Chemical Engineering at Aarhus University, replicated the study and added QIAGEN CLC Genomics Workbench v.23.0.5 (CLC) to the mix, equipped with the Biomedical Genomics Analysis plugin. Specifically, the Immune Repertoire Analysis tool of CLC was used, which was developed for bulk RNA-seq data and also included in the CLC Single Cell Analysis Module. Each single cell was treated as a separate sample. See the complete workflow.
The following dataset types were analyzed:
Real data: Datasets with BCR sequences from actual immune cells (plasmablasts) obtained from earlier studies
Simulated data: Datasets mimicking real-world scenarios with mutations in the BCR genes (heavy and light chains).

The results, detailed here, show that:
When working on BCR reconstruction (or NGS data in general), you want a tool with everything you might need. CLC offers a comprehensive toolset for immune repertoire analysis of single-cell data, among other applications.
You also need a software package that is easy to set up, does not require coding and works across various hardware. There would be no need to invest in new hardware and spend weeks or months learning a programming language. This makes it easier to get started and enables you to generate insights from your data right away. Remember – the right tools can take your research to the next level.
Learn more or request a trial of CLC Genomics Workbench, your all-in-one toolkit.
References:
Author acknowledgments: We thank Dr. Tommaso Andreani, Senior Principal Data Scientist at Sanofi, for continuous support and encouragement during this study.