Infectious diseases impact public health globally. Pathogen outbreaks affect the life of patients and create a high economical burden.
Early detection and control of outbreaks is therefore key to protecting public health.
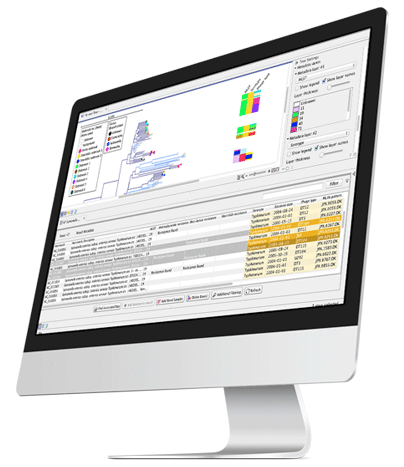
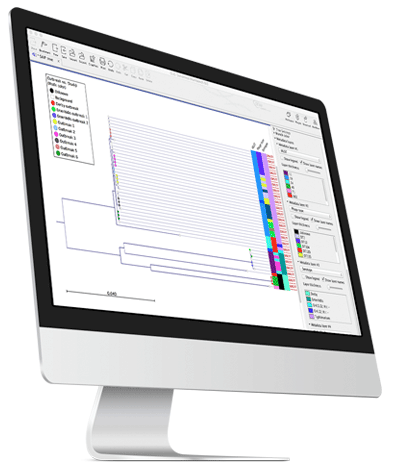
Whole genome analysis offers unique advantages when used for typing and characterizing microbial isolates. Whole genome data reveals the full picture providing taxonomic information and insight into factors like antimicrobial resistance. Once a pathogenic microbe is typed and characterized, comparative whole genome SNP analysis is the most accurate path to tracking a pathogen contamination back to the source.
While crucial for outbreak control, whole genome analysis also enables quality control of starter cultures and biotechnologically valuable strains.
Samples, metadata and all typing results are collected in an Analysis Dashboard providing a powerful overview of outbreak isolates.
A single analysis Workflow for typing returns different types of information for any number of isolates:
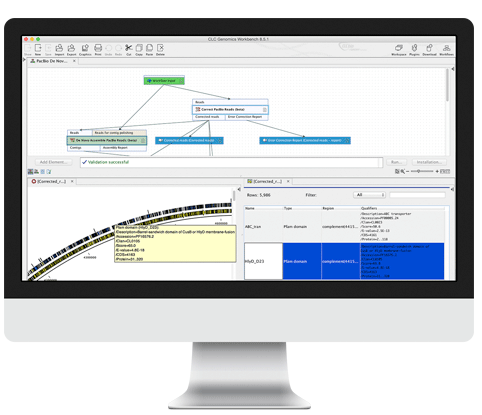
Raw PacBio reads are error-corrected and de novo assembled using a novel
approach that can quickly convert reads to high quality reference genomes.