

















Recent updates to the Whole Genome Alignment plugin for QIAGEN CLC Genomics Workbench feature several improvements to visualizations and functionality to help you more easily gain insights into your microbial genome research.
Here we highlight how these new features improve the Create Whole Genome Alignment tool:
To showcase the improved visualizations, we used the 23 Streptococcus thermophilus circular genomes as used by Alexandrakiet, V. et al. (1) for a study on the evolution and biology of this bacterial species. The genomes are all complete on the chromosome-level, with one ~1.8 Mbp circular scaffold.
Here, we present the whole genome alignments for the Whole Genome Alignment 20.0 beta release (Figure 1) and the updated Whole Genome Alignment 20.1 release (Figure 2).
Both alignments were created using default alignment settings.
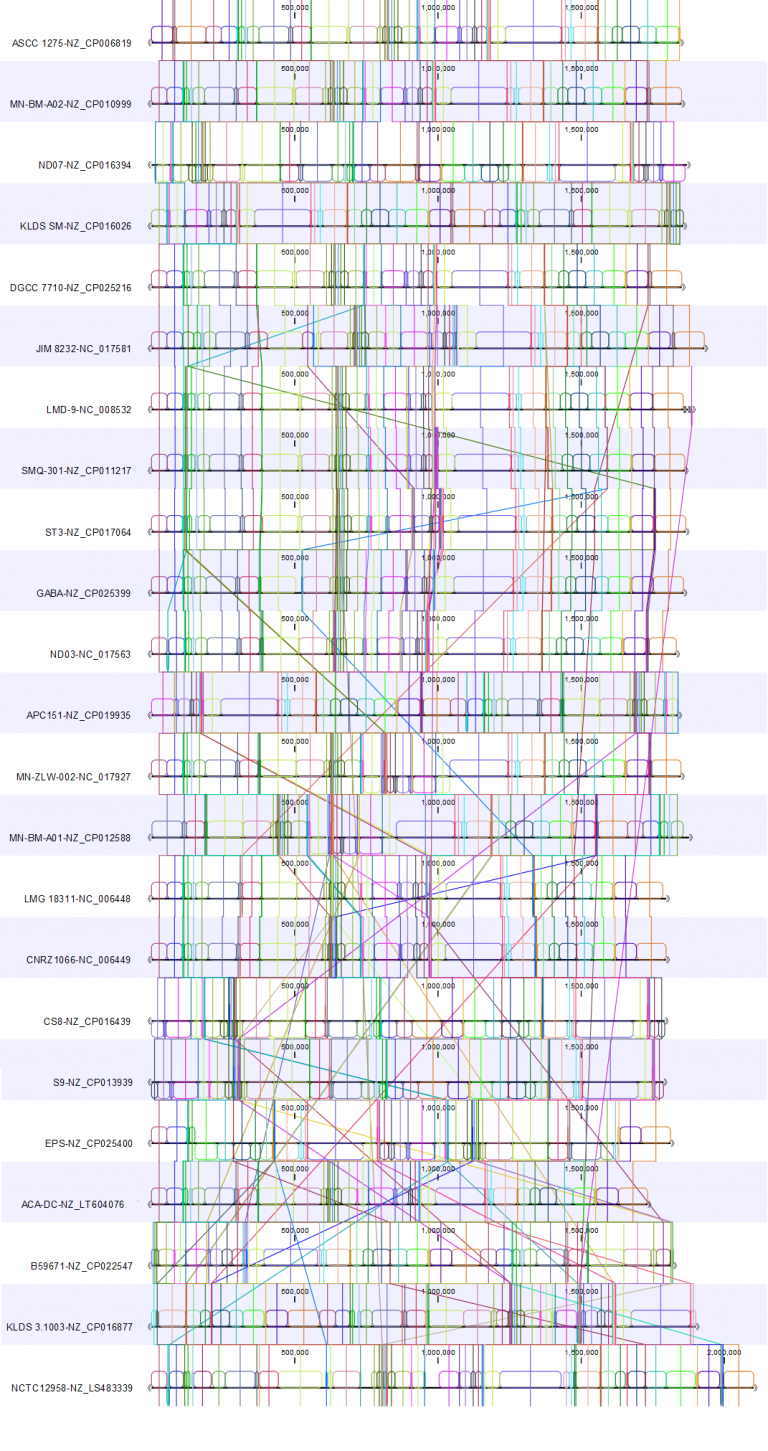
For the 20.1 release, we used the genome from strain KLDS SM (NZ_CP016026.1) as the reference for the rearrangement. In these two alignments, we see how rearranging the genomes to allow reverse complementing and circular shifting relative to the reference results in a cleaner view that allows identification of alignment blocks with minimal crossing. This makes it easy to spot where the genomes differ.
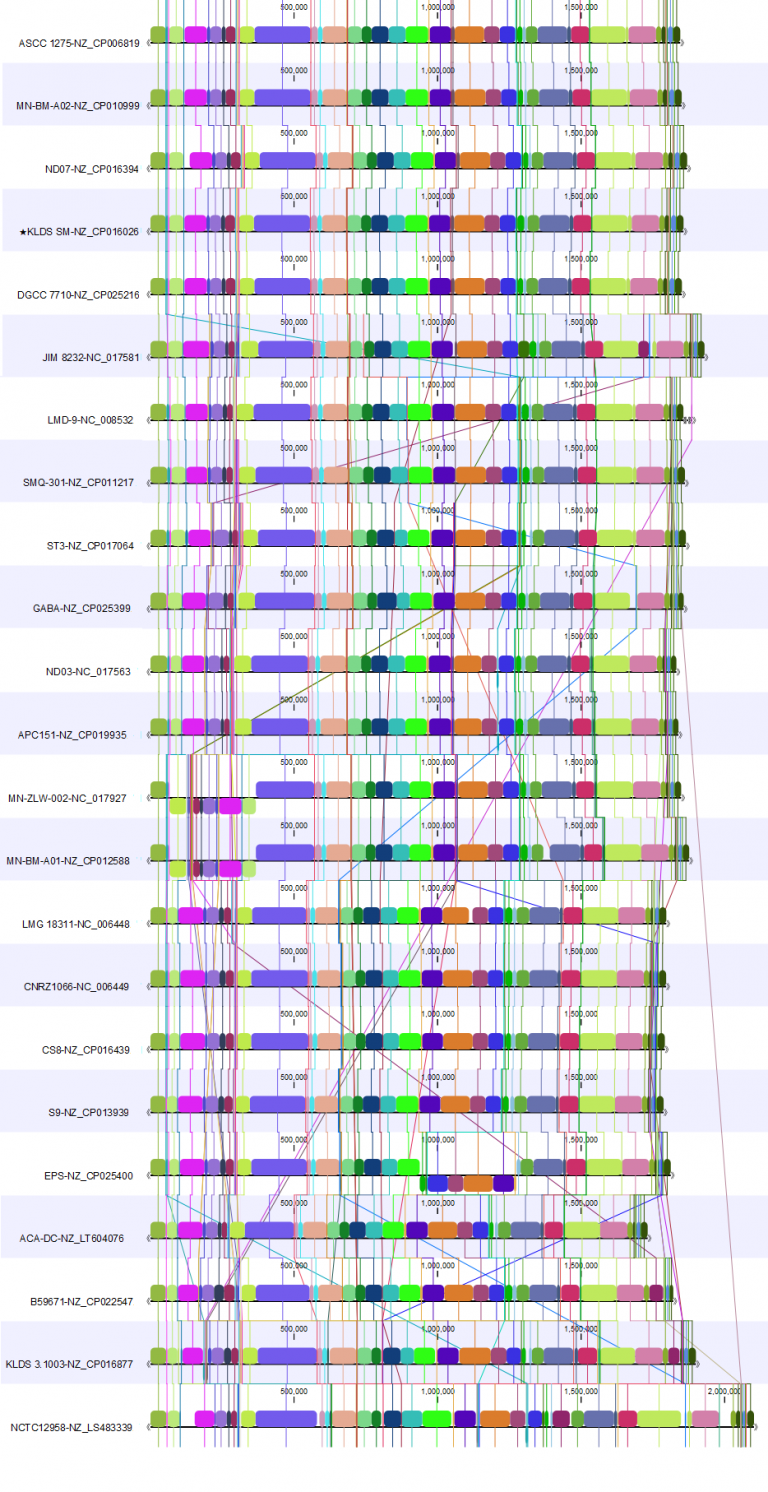
The 20.1 update provides an improved overview of the genomes we are studying. From this, we can conclude that the genomes are complete and similar.
As an example of where to start with an analysis deep-dive, we studied how the reference strain differs from the other selected strains. For this, we utilised the new view Color by reference position (available under Whole Genome Alignment settings) (Figure 3). This creates a color gradient over the reference genomes and colors the alignment block of the other genomes according to their position in the reference genomes. Blocks not found in the reference genome are shown in green.
We have also rearranged the order in which the genomes are presented in the alignment to focus on 3 genomes. Changing the order is possible when Show tree is disabled.
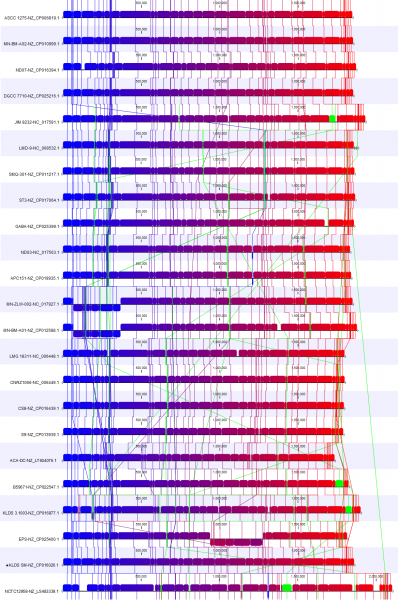
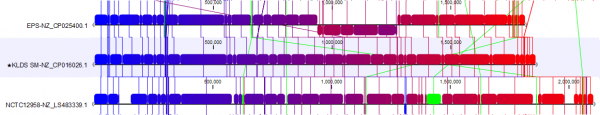
In Figure 3B, we focus on a subset of the alignment. Here, we see that strain NCTCC1958 has a genome stretch of around 60 Kbp (shown in green) which is not present in the reference genome. To explore this further, we can enable annotations in Annotation layout. By zooming in and mousing-over, we can see this alignment block covers several genes (Figure 4).
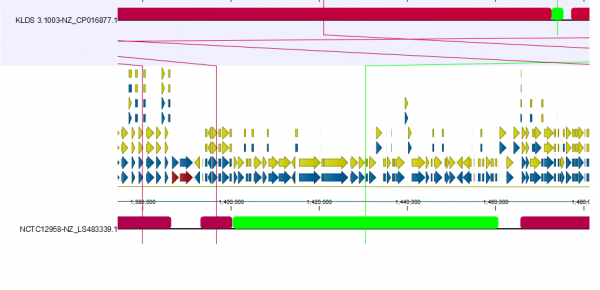
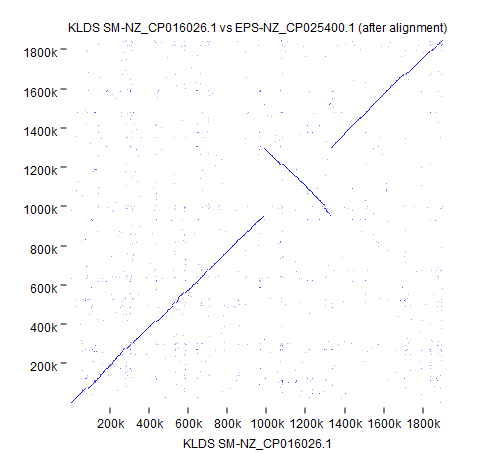
The other selected strain, EPS (NZ_CP025400.1), has a 300 Kbps inversion that is now easy to spot thanks to circular shifting and the color gradient.
Using the genome output after alignment, we can confirm this inversion with Create Whole Genome Dot Plot using default settings (Figure 5).
Here we describe only a subset of the new features in the Whole Genome Alignment 20.1 tool of QIAGEN CLC Genomics Workbench. To explore the full capabilities of QIAGEN CLC Whole Genome Alignment tools, see:
References:












