
















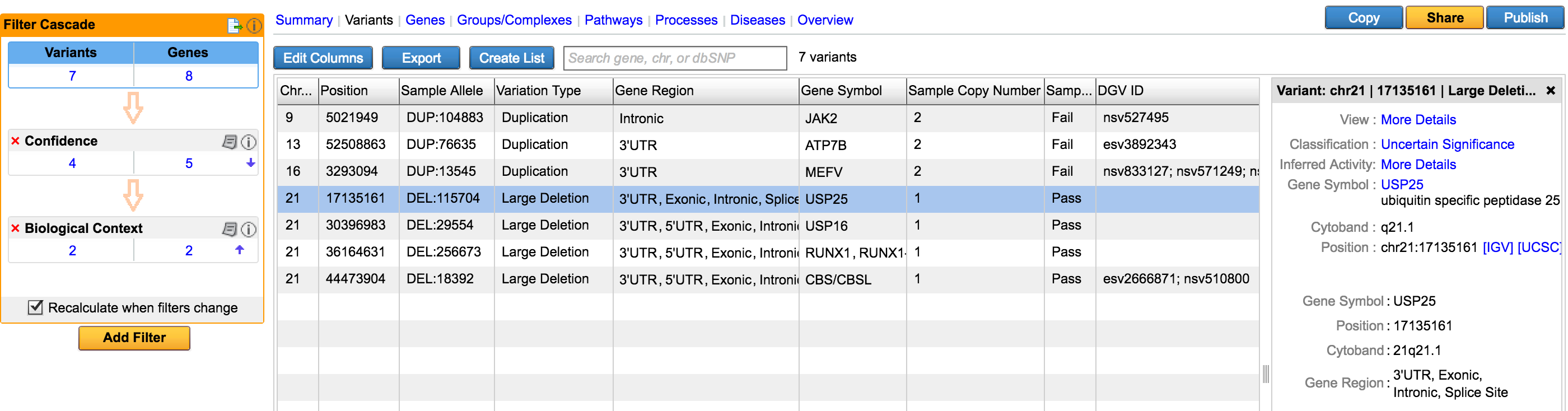
Support Copy Number Variations from VCF
Copy Number Variations have long been associated with inherited disease predisposition, as well as cancer initiation and development. With the decreasing cost of WGS and increased accuracy of secondary analysis algorithms, more NGS workflows are taking an integrated approach to variant detection that spans SNVs, small indels, and large-scale genomic alterations. Variant Analysis can now accept deletion and duplication structural variants specified in the VCF input. To assess the phenotypic effect of these variants and their role in disease, users have at their disposal a number of filtering and annotation capabilities to perform common CNV analysis tasks. These include removal of spurious calls, overlap with genes affected, comparison to known CNVs in healthy populations, and co-occurence with other variants as well as across multiple samples.
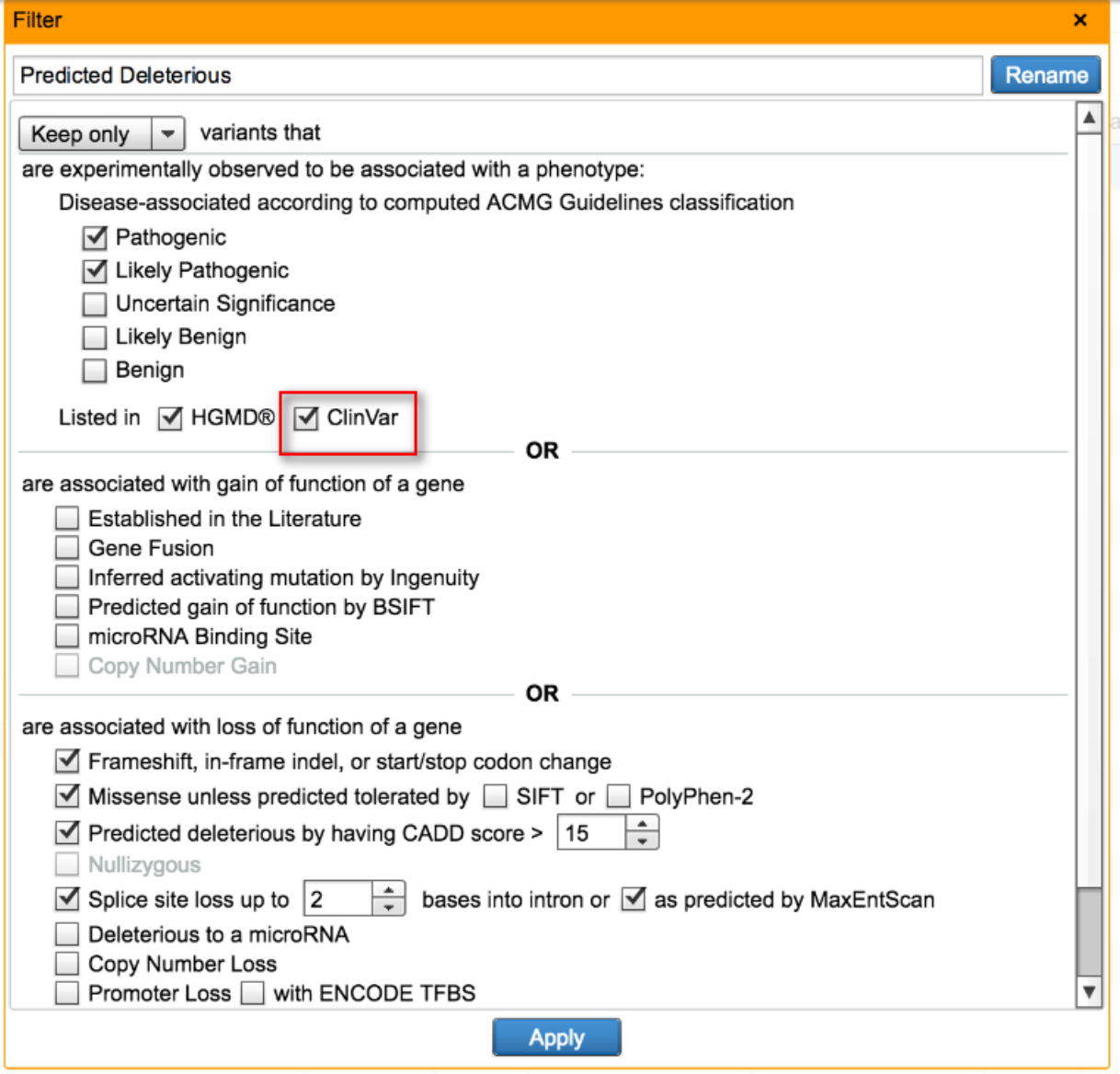
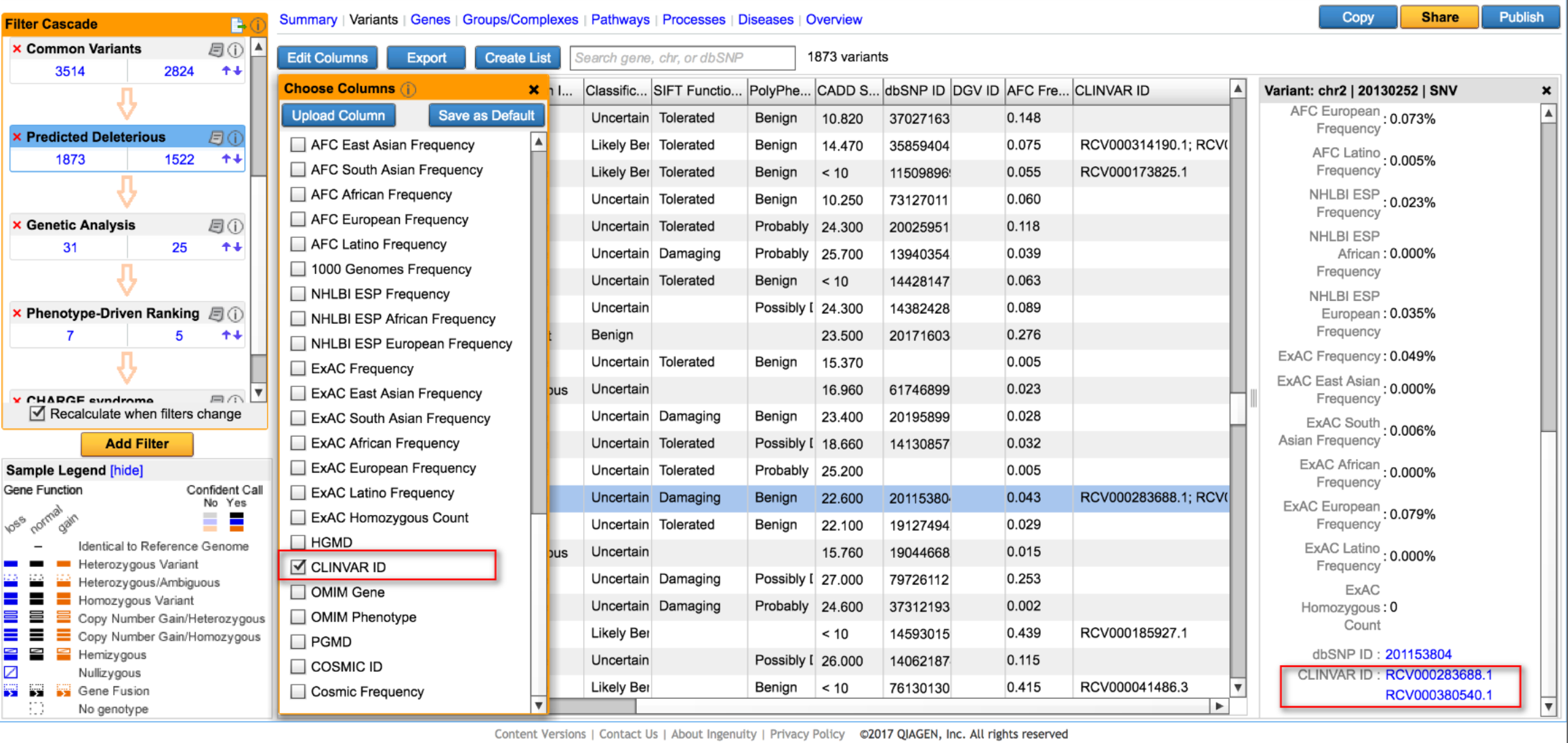
Filter and annotate variants using ClinVar
In the Predicted Deleterious Filter, in addition to leveraging QIAGEN computed ACMG classifications and presence in HGMD, users can now check for presence in ClinVar as another means to identify and retain disease-associated variants. For variants that are present in ClinVar, they will be annotated with ClinVar accession IDs and link-outs, multiple if more than one condition is associated with that variant.

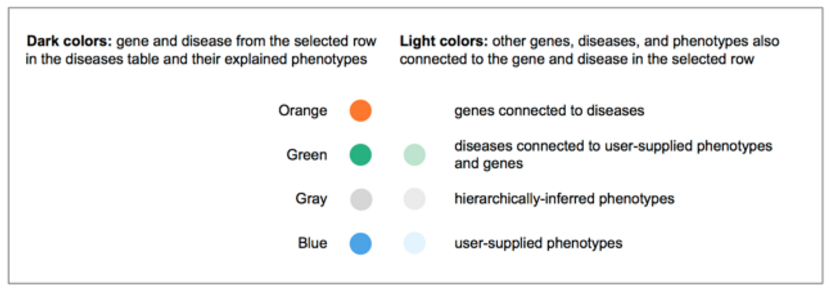
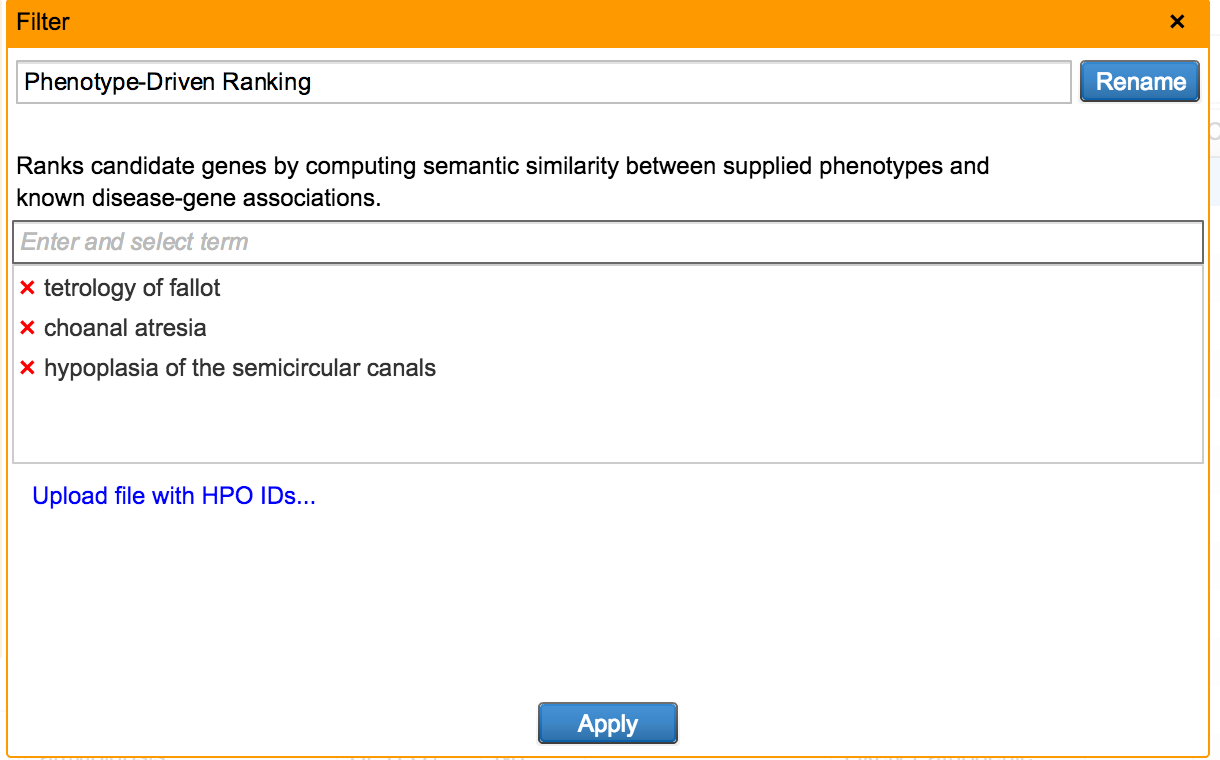
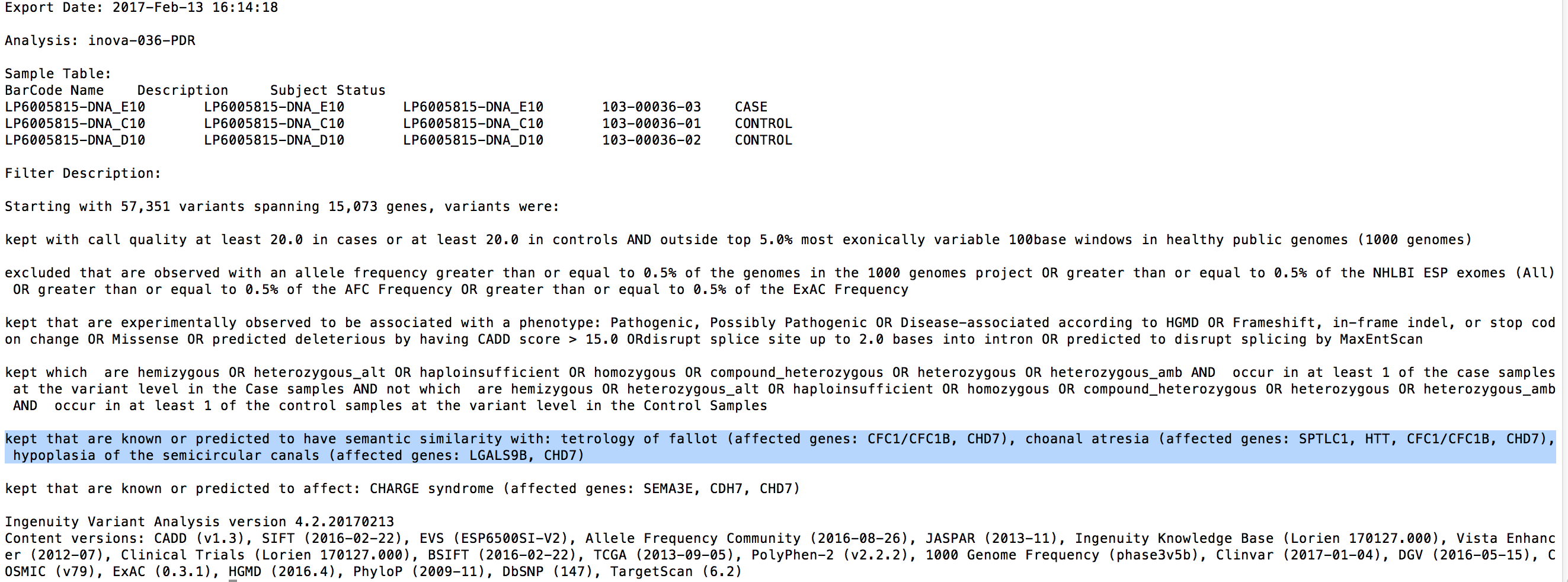
Content versions: CADD (v1.3), EVS (ESP6500SI-V2), Allele Frequency Community (2017-01-31), JASPAR (2013-11), Ingenuity Knowledge Base (Lorien 170127.000), Vista Enhancer (2012-07), Clinical Trials (Lorien 170127.000), BSIFT (2016-02-23), TCGA (2013-09-05), PolyPhen-2 (v2.2.2), 1000 Genome Frequency (phase3v5b), Clinvar (2017-01-04), DGV (2016-05-15), COSMIC (v79), ExAC (0.3.1), HGMD (2016.4), PhyloP (2009-11), DbSNP (149), TargetScan (6.2), SIFT4G (2016-02-23)
Note: The new features and improvements mentioned above will be available on Saturday 25th March, as soon as the release-related system maintenance is completed.













