
















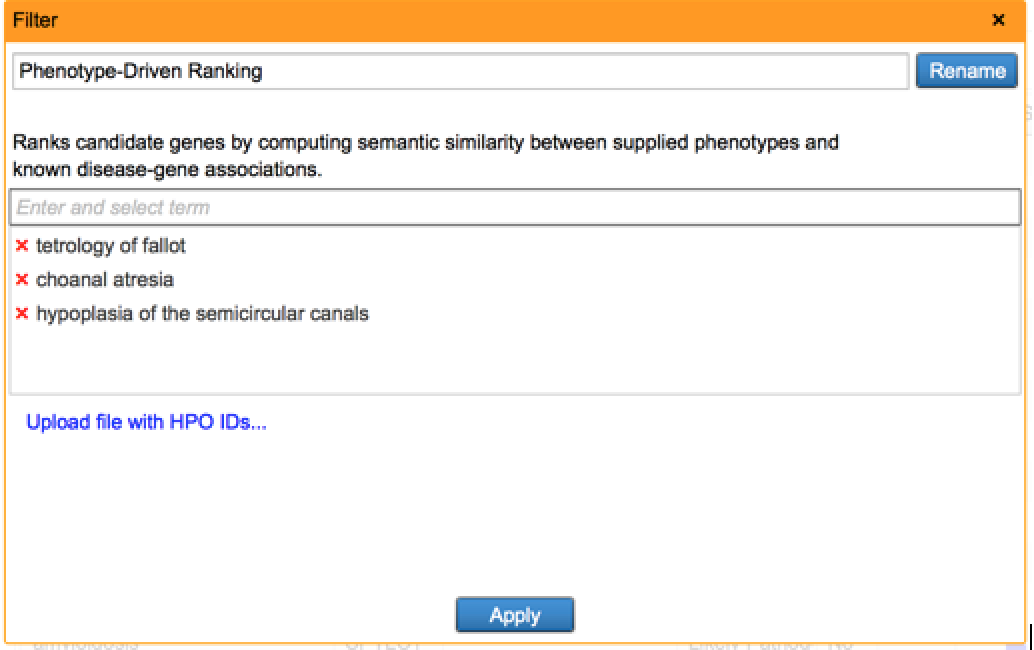
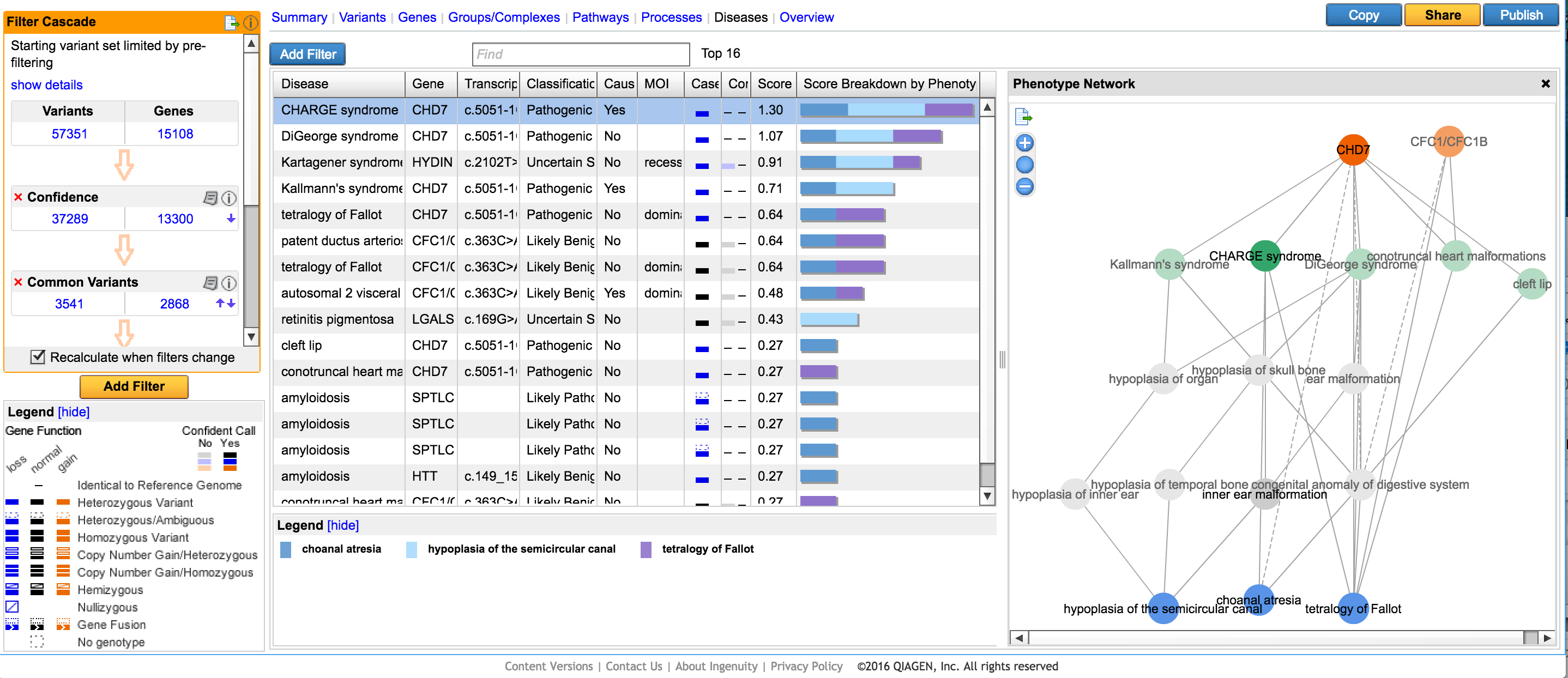
Expedite disease discovery using Phenotype-Driven Ranking filter
This new filter prioritizes and ranks variants by using user-supplied phenotype and genotype data in conjunction. This approach draws from a network of phenotype-phenotype, phenotype-disease, and disease-gene relationships established from the QIAGEN Knowledge Base, and looks for plausible diseases that can explain both the phenotypes observed as well as the genetic variations detected. For each disease, we can compute a score that represents the compatibility between the phenotype profile and disease, and this score is in turn used to rank variants which reside in disease-implicated genes. Read more about this filter in our white paper.
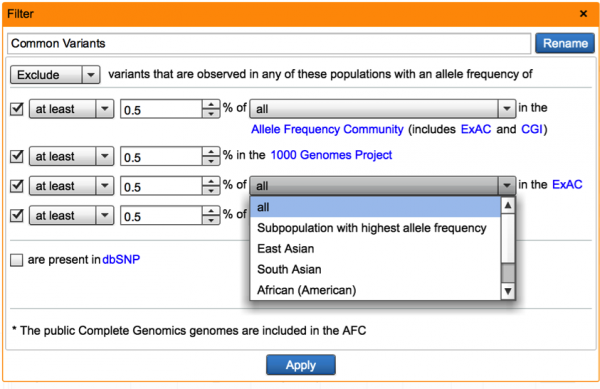
Evaluate variants using subpopulation Allele Frequency from ExAC
In addition to filtering with aggregate frequency, users can now filter variants by their associated frequencies in ExAC ethnic subpopulations (East Asian, South Asian, African American, European, Latino), including a max-population frequency to ensure that the variant frequency is sufficiently low in each population. The max-population frequency is also incorporated into ACMG rule calculations where ExAC is used as a basis for controls data or common/benign variants. Similarly, we have added a max-population frequency option for filtering using the Allele Frequency Community dataset. For mendelian disorders, one expects a highly-damaging, causal variant to be rare across every population, so the ability to leverage multiple ethnicities in filtering and ascribing clinical significance is of high utility.
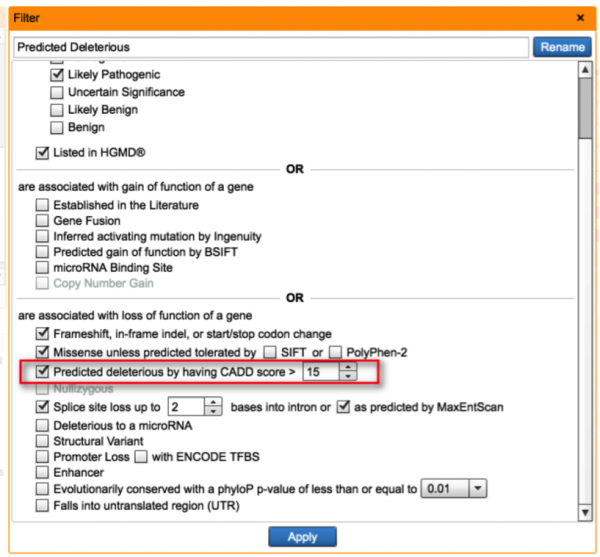
Incorporate CADD scores for functional predictions
Based on published literature comparing score distributions of several in-silico functional prediction approaches, CADD not only covers intronic and exonic regions, but also produces better separation between causal and benign variants than independent metrics like SIFT and PolyPhen. There are a number of well-established pathogenic variants where SIFT and PolyPhen predicts the variant to be benign, so the ability to use another prediction tool lowers the risk of discarding potentially important variants when establishing criteria for loss-of-function in the Predicted Deleterious filter.
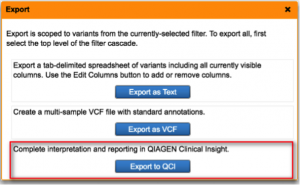
Streamline Variant Analysis to QCI Interpret workflow
QCI Interpret is a clinical decision support solution designed for genetic testing labs using next-generation sequencing platforms, catering to both somatic and hereditary cancer interpretation needs. It provides ACMG-guided variant classifications and rationale, clinical case counts from curated literature, as well as eligible treatments and clinical trials. By offering Variant Analysis users the ability to export filtered set of variants directly to QCI Interpret along with relevant metadata, we bridge the gap between variant filtering and variant reporting for clinical test offerings that involve large panels, exomes, or genomes. Prior to QCI Interpret, users can also supply data collected from additional family members or normal tissue for comparisons between case and control samples. Using the two products in unison brings efficient reduction of the candidate variant list in Variant Analysis, followed by fine-grained assessment and configurable reporting of actionable variants in QCI Interpret.
To learn more about QCI Interpret and request a demo, please visit the QCI Interpret product page.
Improvements
Ingenuity Variant Analysis version 4.2.20160927
Content versions: CADD (v1.3), SIFT (2016-02-22), EVS (ESP6500SI-V2), Allele Frequency Community (2016-08-26), JASPAR (2013-11), Ingenuity Knowledge Base (Jakku 160913.000), Vista Enhancer (2012-07), Clinical Trials (Jakku 160913.000), BSIFT (2016-02-22), TCGA (2013-09-05), PolyPhen-2 (v2.2.2), 1000 Genome Frequency (phase3v5b), Clinvar (2016-06-01), COSMIC (v77), ExAC (0.3.1), HGMD (2016.2), PhyloP (2009-11), DbSNP (147), TargetScan (6.2)













