
















The issue described here affects results of the tools Quantify QIAseq UPX 3′ and Analyze QIAseq Panels guide -> UPX 3′ RNA of the Biomedical Genomics Analysis plugin when used on QIAGEN CLC Genomics Workbench 20.0 or 20.0.1.
If you are using affected tools, please upgrade your software. This issue was fixed in QIAGEN CLC Genomics Workbench 20.0.2 and QIAGEN Genomics Server 20.0.2. See the Affected software and tools section below for further details.
To check the software version used to generate a data element, you can refer to its history information.
The QIAseq UPX 3′ Transcriptome protocol involves two amplification steps, one before and one after fragmentation:
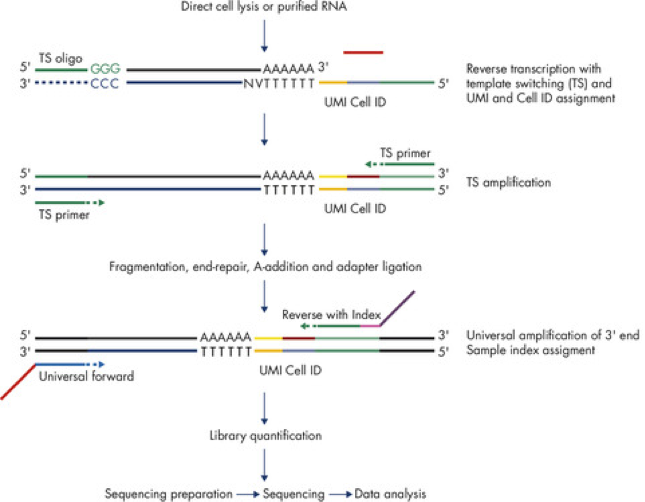
When quantifying expression, affected software only uses UMIs to correct for duplicate molecules generated during the second amplification step. It does not correct for duplicate molecules from the first amplification step.
This issue leads to systematically high expression values for affected samples.
Samples are most likely to be affected when the sequencing depth is high compared to the amount of input RNA. For example, most single-cell data is likely to be affected.
Any applications making direct use of expression values (including TPM and RPKM and absolute counts) will be affected.
Where effects are normalized: It is likely that overall results will not be strongly affected for analyses involving normalization. For example, the most significant differentially expressed genes of an analysis are likely to be correctly detected despite this issue.
QIAseq UPX 3′ Targeted RNA Panels: Analysis of data from these panels is largely unaffected by this issue. However, for some panels, there is a possibility that the same molecule will be amplified by two different primers. In such cases the resulting duplicate molecules will not be detected due to the issue described here.
This issue affects the tools Quantify QIAseq UPX 3′ and
Analyze QIAseq Panels guide -> UPX 3′ RNA, delivered by the following plugins:
This issue was fixed in CLC Genomics Workbench 20.0.2 and CLC Genomics Workbench 20.0.2, where choosing the “3′ sequencing” Library type setting of RNA-Seq Analysis when analyzing reads that have been annotated with UMIs by tools of the Biomedical Genomics Analysis plugin, results in expression values in the GE track being based on the number of distinct UMIs for each gene, rather than the number of reads.













