
Latest improvements for Ingenuity Variant Analysis
Ingenuity Variant Analysis (IVA) Spring 2019 Release
Now you can include even more metadata and variant types in your IVA analyses
The IVA Spring 2019 Release offers expanded analyses parameters to improve your analyses. As of April 14, 2019, IVA includes the following new features:
Updated VCF parser to capture additional upstream metadata
IVA will now parse more information from VCF headers to capture experiment and sequencing run metadata. Specific header formats must conform to the following format where ##<header tag> -> is followed by pipeline metadata:
##fileDate -> ING:variantFileDate
##source -> ING:variantFileSource
##workflow_type -> ING:workflowType
##workflow_version -> ING:workflowVersion
##cmdline -> ING:variantCmdLine
For CGI files the following header tags are supported:
#GENERATED_BY -> ING:variantFileSource
#GENERATED_AT -> ING:variantFileDate
Updated VCF parser to capture additional variant types
While IVA has always supported CNV, it will now also support fusions. This requires:
##INFO=<ID=SVTYPE,Number=1,Type=String,Description="Type of structural variant">
Where Type of structural variant includes (case sensitive):
Fusion – fusion
BND – breakend
DUP - duplication
DEL – deletion
Please consult IVA documentation here for further details
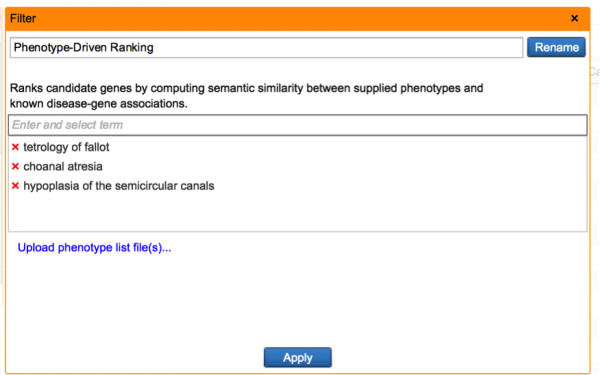
Phenotype driven ranking now an option in the Analysis Creation Wizard
We have now provided the option to select the Phenotype Driven Ranking as part of the Analysis creation. This option is only available where valid phenotype terms are applied in the Biological Context box.

Deprecation of hg18 from IVA experts US-based IVA
In an effort to better server IVA users world-wide, we have removed support for hg18 on IVA datacenters outside the USA. This will increase performance of non-USA datacenters. If you plan to upload samples based on the hg18 human reference, please contact Support to ensure you are uploading such samples to the USA-based IVA datacenter.
Updated content
The IVA Spring 2019 Release contains the following content:
- HGMD v2018.4
- PGMD v2018.1
- DbSNP v151
- AFC v2018-05-22
To view the latest version of the underlying QIAGEN Knowledge Base, please click on the Content Versions link at the bottom of the page.

For more information about IVA and the Spring 2019 Release, visit our Resources page.
Winter 2018 Release
Filtering for CentoMD® variants and easily tracking sample uploads!
We are pleased to announce that the Fall 2018 Release of IVA has arrived, and with greater functionality and improved data analysis export.
You can now:
Filter for CentoMD variants within the Predicted Deleterious filter

Export variants with Sample IDs automatically included
IVA now includes the Sample IDs when you export the variants table.This feature is only available in the Export to Text option.



Easily track the available balance of sample uploads in your subscription
You can now see the available balance of your subscription upload limit. This new notification will show you how many more samples you can upload to your IVA account based on your subscription license and end date.
Obtain email receipts for each sample


To confirm your 'purchase' of an uploaded sample(s), IVA now requires confirmation of sample upload. This gives you the option to review and confirm the sample you wish to upload. In addition, you can choose to receive an email 'receipt' of your sample upload. Should you have insufficient upload credits, your attempted samples upload will be rejected and you will be prompted to upload fewer samples.
Email notification of samples upload and validation. Starting with this release, IVA will send email confirmation of your sample upload and validation. These emails will act as a record of your sample uploads purchase.
To view the latest version of the underlying QIAGEN Knowledge Base, please click on the Content Versions link at the bottom of the page.
Check out the full release summary here!
Summer 2018 Release
Tighter integration with QIAGEN Clinical Insight (QCI)
In this release, there will be a new “Application” column under the “My Samples” and “My Analyses” tabs. Users with an IVA and QCI account will now be able to clearly see which samples or analyses belong to which application.
Samples are considered QCI samples under the following conditions:
- Sample(s) are exported from IVA to QCI.
- Sample(s) are uploaded directly via QCI.
If a sample is labeled as a QCI sample, it cannot be deleted. The "Delete" button will be greyed out to prevent removal of QCI samples.
To remove QCI samples from tabular view, select "Settings" in the upper-right corner and uncheck the “Show QCI samples and analyses” option. You will still be able to see QCI samples that are associated with IVA, which are samples that have been initially uploaded via IVA, then exported to QCI.
Updated content
In this release, the following content has been added:
- HGMD v2018.1
- PGMD v2018.1
- dbSNP v151
- AFC v2018-05-22
To view the latest version of the underlying QIAGEN Knowledge Base, select "Content Versions" at the bottom of the page.

Updated gene model
We've updated the underlying gene model to improve the accuracy of handling HGVS right-shifting and to support ENSEMBL transcripts using Genecode (v27) from ENSEMBL v90 and v91.
Spring 2018 Release
Improved Data Export
With this new release, export or download of data from IVA will be performed offline. That is, rather than being able to download at the time of exporting, you will now receive an email with a link to download your exported file(s). This improves performance and allows the resuming of interrupted downloads.
When you export, you receive a pop-up message. Once you receive your email notification that your download is ready, you may click on the link to download. Note that you will only have 30 days to download the file before the link becomes inactive. The downloaded files will be compressed in gzip format. If you have problems uncompressing gzip formats, we recommend using the free utility 7-Zip
Improved Handling of Uploaded VCF Files
In the past, uploaded VCF files would fail if the vcf file was missing FORMAT or GT values. This update improves processing of VCF files to allow files lacking FORMAT or GT to remain valid for downstream processing and analysis.
Addition of CentoMD Data into the AFC
This release also features new CentoMD data added to the Allele Frequency Community (AFC). The CentoMD data set contains over 155,000 sequenced individuals who’s genotype will now be incorporated into the AFC for a more comprehensive view of population genetics.
Updated Allele Frequency Community Statistics
The Allele Frequency Community (AFC) contain summary statistics on alleles contributed from public, private, and QIAGEN community users. The statistics are now based on over 750,000 samples analyzed through the QIAGEN bioinformatics platform including over 38,000 whole genomes and over 358,000 exome samples.
Winter 2017 Release
Support Copy Number Variations from VCF
Copy Number Variations have long been associated with inherited disease predisposition, as well as cancer initiation and development. With the decreasing cost of WGS and increased accuracy of secondary analysis algorithms, more NGS workflows are taking an integrated approach to variant detection that spans SNVs, small indels, and large-scale genomic alterations. Variant Analysis can now accept deletion and duplication structural variants specified in the VCF input. To assess the phenotypic effect of these variants and their role in disease, users have at their disposal a number of filtering and annotation capabilities to perform common CNV analysis tasks. These include removal of spurious calls, overlap with genes affected, comparison to known CNVs in healthy populations, and co-occurence with other variants as well as across multiple samples.

Filter and Annotate Variants Using ClinVar
In the Predicted Deleterious Filter, in addition to leveraging QIAGEN computed ACMG classifications and presence in HGMD, users can now check for presence in ClinVar as another means to identify and retain disease-associated variants. For variants that are present in ClinVar, they will be annotated with ClinVar accession IDs and link-outs, multiple if more than one condition is associated with that variant.


Improvements
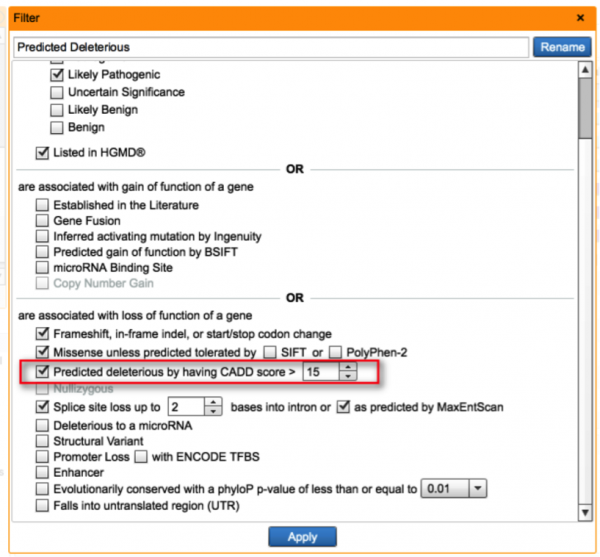
- For functional prediction of variants, complete integration of CADD scores by bringing in CADD scores for known insertion and deletion variants, in addition to SNVs.
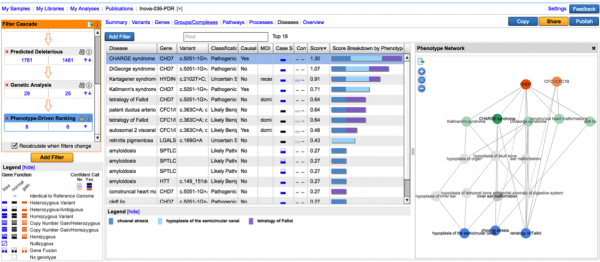
- Increase flexibility and readability of Phenotype-Driven Ranking Filter (PDR) network graph display by allowing users to toggle between highlighted nodes only vs. all nodes. Highlighted nodes reflect genes and diseases from the selected row in the Diseases tab, whereas related nodes reflect diseases and phenotypes connected to the gene and disease in the selected row.



- In the table presented in PDR Filter -> Diseases tab, utilize PDR disease context for computing the variant’s ACMG classification rather than showing default ACMG classification (most pathogenic).
- Provide list of genes implicated when exporting filter cascade settings for terms entered into the Biological Context and Phenotype-Driven Ranking filters.


- Use a more accurate calculation for allele fraction in the case of Ion Torrent VCFs (AF = FAO/FAO + FRO).
- Allow special characters (' $ % ^ &) to be used in filter naming conventions, which can be part of QIAGEN KB disease names.
Content versions: CADD (v1.3), EVS (ESP6500SI-V2), Allele Frequency Community (2017-01-31), JASPAR (2013-11), Ingenuity Knowledge Base (Lorien 170127.000), Vista Enhancer (2012-07), Clinical Trials (Lorien 170127.000), BSIFT (2016-02-23), TCGA (2013-09-05), PolyPhen-2 (v2.2.2), 1000 Genome Frequency (phase3v5b), Clinvar (2017-01-04), DGV (2016-05-15), COSMIC (v79), ExAC (0.3.1), HGMD (2016.4), PhyloP (2009-11), DbSNP (149), TargetScan (6.2), SIFT4G (2016-02-23)
Note: The new features and improvements mentioned above will be available on Saturday 25th March, as soon as the release-related system maintenance is completed.
Fall 2017 Release
Improved Phenotype Driven Ranking of Candidate Variants
With this release, variants that have the same Phenotype Driven Ranking (PDR) score will now be further re-ranked using a tie-breaking logic. Diseases are rank-ordered first by score (high scores first), then as a tie-breaker by gene-disease causality (causal first), then disease prevalence (higher prevalence first), and finally alphabetical order.
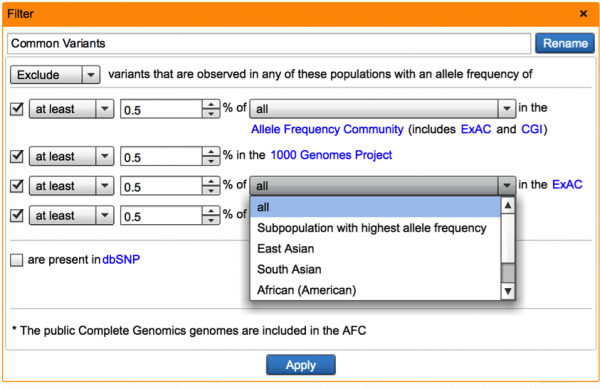
Allow Established Pathogenic Variants to Pass the Common Variants Filter
In certain situations, there are a set of common variants that are report as pathogenic despite having greater than 1% population allele frequency.
While we typically want to exclude all variants above 1% otherwise, this creates a challenge when applying the Common Variants filter with 1% which will exclude these important pathogenic mutations when they occur, and raising the CV filter to 4% or higher includes too many irrelevant (non-pathogenic) variants that slows down review time.
This new improvement allows VA to be able to filter out variants above a % allele frequency threshold while keeping a short list of known common pathogenic variants. Generally these are 'founder' variants in the sense that they occurred recently in human evolution at a population bottleneck causing many people to inherit the mutation from that one affected founder. To enable this feature, you'll need to check the "unless Established Pathogenic Common variant”.

Improved User Experience To Clarify The Range of Bases Defining a Gene
A Biological Context (BC) filter, filters for variants based on known genes derived from the BC terms entered in the BC filter. However the scope of a gene can be modified through the Predicted Deleterious (PD) filter when the user specifies how many bases into the intron (from the intron/exon border) one wants to retain. For example to consider a gene ± 10 bases, one can specify "Keep only variants no more than 10 bases into intron...". In this case, the BC filter will include variants within the gene's exons as well as intronic variants within 10 bases from the exon's border.
The improved UI of the PD filter makes this clearer and less confusing. If a user wants to consider a gene ± 10 bases, one can specify "Keep only variants no more than 10 bases into intron...". The downstream BC filter will now consider variants within relevant genes including variants found within the gene's exons as well as ± bases beyond the exon's border. If one wants to consider possible variants in all intronic locations, one should not select this option.

You Can Now Cancel of Resume the Opening of an Analysis
Occasionally you open a large analysis that takes some time to open. In the middle of opening such an analysis, if the browser closes, changes webpage, or the computer is shutdown, an attempt to re-open such an analysis would have resulted in a warning with no option to cancel or resume the opening of the large analysis. With this new release, you're now able to resume or cancel such a large analysis which was previously interrupted.

Improved BED File Handling and Presentation of BED File Data
This latest release bow support/show correctly in the analysis the negative values from the BED files and also support more precision (decimals) for custom fields.
Summer 2017 Release
New gnomAD database for filtering Common Variants
We're very excited to announce the availability of the gnomAD dataset in Ingenuity Variant Analysis. Within the Common Variants filter there is now a new population to filter common variants:

gnomAD is the second iteration of the popular ExAC content with the addtion of population genomes and exomes. The dataset contained in gnomAD consists of 123,136 exomes and 15,496 genomes. More information on gnomAD can be found at the gnomAD website.
New Centogene Variant Annotations
Centogene's CentoMD database is the world's largest rare disease mutation database which combines clinical genetic information with actual clinical patient case data. This database is rich with information including genotype-phenotype data on not-yet-published variants leading to even more evidence-driven insights. If a variant has CentoMD annotations, it will display a CentoMD link in the properties window. Clicking on the link will link out to the CentoMD website to display additional Centogene annotations. To view the additional Centogene annotations a subscription to CentoMD is required.


Ashkenazi Jewish subpopulation is now available
Given that gnomAD contains 5000+ Ashkenazi Jewish individuals, this release provides a subpopulation frequency for Ashkenazi Jewish if filtering with gnomAD within the Common Variants filter.

My Filters
Introducing My Filters. You can now save your cascade filtering settings under my filters that you can access via the new My Filters tab:

Within the My Filters tab you will see your collection of saved filters that you can apply to existing and future IVA analysis. Also, if you have access to QIAGEN Clinical Insights (QCI) you can make these filters available to QCI for pre-filtering (by IVA) then exporting the filtered variants to QCI for reporting. Saved filters can be shared with other IVA users.
To create a filter simply click the floppy disk icon at the top of the filter cascade:

To save the filter cascade settings. You'll need to name your filter setting and optionally a brief description:

To see all your saved filters, click on the My Filters tab:

To apply you saved filter to a new analysis on the pre-configurator window:

Streamlined Export of Filters to QIAGEN Clinical Insight
After optimizing filter settings in Variant Analysis, such filters can be saved in My Filters which are also available as Pre-filter Settings within QIAGEN Clinical Insights (QCI). Now QCI users can upload large VCF files directly within QCI (exome/genomes) and select a list of pre-filters from the user's Variant Analysis' My Filters library. This saves the QCI user from having to first upload via Variant Analysis, pre-filter, then export to QCI.
Support for HPO Terms in the Biological Context Filter
You can now directly enter HPO terms in the Biological Context filter rather than adding biological terms. Simply upload a text file containing a list of HPO terms using the Upload file(s) link within the Biological Context filter.

Improved Error Messaging for VCF File Upload Failures
When customers experience a failed VCF upload, having useful error message will help users troubleshoot the upload problems with the VCF file. Additionally, users in this release of the Variant Analysis API will experience richer error messages in the event of an empty or invalid sample and have the ability to request further details and counters regarding the sample.


Improved Indel Handling in Allele Frequency Community (AFC)
Normalize and left-align all indels in user opted-in datasets in AFC while incorporating gnomAD into AFC.
Spring 2017 Release
Improved Indel handling
Normalize indels in dbSNP to ensure that all short insertion/deletions are left-aligned, such that correctly formatted variants in user VCF data will be consistently annotated with the corresponding dbSNP record
Streamlined request to toggle pre-filtering
Add ability to dynamically handle and respond to customer requests for disabling pre-filtering for configuring analyses. Customers can initiate the request to Variant Analysis Support Team, who will work with product team members to turn off/turn on pre-filtering as a per-user setting for the duration that is required.
New beta feature – Set Variant Assessment via API
For high volume labs who want to set their own variant assessments using the QCI’s application programming interface (API). This feature allows for bulk upload/updates of QCI’s variant assessments via the API. This feature requires the API License.
New features in the Winter 2017 Release
Support Copy Number Variations from VCF
Copy Number Variations have long been associated with inherited disease predisposition, as well as cancer initiation and development. With the decreasing cost of WGS and increased accuracy of secondary analysis algorithms, more NGS workflows are taking an integrated approach to variant detection that spans SNVs, small indels, and large-scale genomic alterations. Variant Analysis can now accept deletion and duplication structural variants specified in the VCF input. To assess the phenotypic effect of these variants and their role in disease, users have at their disposal a number of filtering and annotation capabilities to perform common CNV analysis tasks. These include removal of spurious calls, overlap with genes affected, comparison to known CNVs in healthy populations, and co-occurence with other variants as well as across multiple samples.
Filter and annotate variants using ClinVar
In the Predicted Deleterious Filter, in addition to leveraging QIAGEN computed ACMG classifications and presence in HGMD, users can now check for presence in ClinVar as another means to identify and retain disease-associated variants. For variants that are present in ClinVar, they will be annotated with ClinVar accession IDs and link-outs, multiple if more than one condition is associated with that variant.
Improvements
- For functional prediction of variants, complete integration of CADD scores by bringing in CADD scores for known insertion and deletion variants, in addition to SNVs.
- Increase flexibility and readability of Phenotype-Driven Ranking Filter (PDR) network graph display by allowing users to toggle between highlighted nodes only vs. all nodes. Highlighted nodes reflect genes and diseases from the selected row in the Diseases tab, whereas related nodes reflect diseases and phenotypes connected to the gene and disease in the selected row.
- Provide list of genes implicated when exporting filter cascade settings for terms entered into the Biological Context and Phenotype-Driven Ranking filters.
- Use a more accurate calculation for allele fraction in the case of Ion Torrent VCFs (AF = FAO/FAO + FRO).
- Allow special characters (' $ % ^ &) to be used in filter naming conventions, which can be part of QIAGEN KB disease names.
Content versions: CADD (v1.3), EVS (ESP6500SI-V2), Allele Frequency Community (2017-01-31), JASPAR (2013-11), Ingenuity Knowledge Base (Lorien 170127.000), Vista Enhancer (2012-07), Clinical Trials (Lorien 170127.000), BSIFT (2016-02-23), TCGA (2013-09-05), PolyPhen-2 (v2.2.2), 1000 Genome Frequency (phase3v5b), Clinvar (2017-01-04), DGV (2016-05-15), COSMIC (v79), ExAC (0.3.1), HGMD (2016.4), PhyloP (2009-11), DbSNP (149), TargetScan (6.2), SIFT4G (2016-02-23)
Note: The new features and improvements mentioned above will be available on Saturday February 25, as soon as the release-related system maintenance is completed.
Ingenuity Variant Analysis Fall 2016 Release
What’s New in the Ingenuity Variant Analysis Fall Release (2016)
Expedite disease discovery using Phenotype-Driven Ranking filter
This new filter prioritizes and ranks variants by using user-supplied phenotype and genotype data in conjunction. This approach draws from a network of phenotype-phenotype, phenotype-disease, and disease-gene relationships established from the QIAGEN Knowledge Base, and looks for plausible diseases that can explain both the phenotypes observed as well as the genetic variations detected. For each disease, we can compute a score that represents the compatibility between the phenotype profile and disease, and this score is in turn used to rank variants which reside in disease-implicated genes. Read more about this filter in our white paper.
Evaluate variants using subpopulation Allele Frequency from ExAC
In addition to filtering with aggregate frequency, users can now filter variants by their associated frequencies in ExAC ethnic subpopulations (East Asian, South Asian, African American, European, Latino), including a max-population frequency to ensure that the variant frequency is sufficiently low in each population. The max-population frequency is also incorporated into ACMG rule calculations where ExAC is used as a basis for controls data or common/benign variants. Similarly, we have added a max-population frequency option for filtering using the Allele Frequency Community dataset. For mendelian disorders, one expects a highly-damaging, causal variant to be rare across every population, so the ability to leverage multiple ethnicities in filtering and ascribing clinical significance is of high utility.
Incorporate CADD scores for functional predictions
Based on published literature comparing score distributions of several in-silico functional prediction approaches, CADDnot only covers intronic and exonic regions, but also produces better separation between causal and benign variants than independent metrics like SIFT and PolyPhen. There are a number of well-established pathogenic variants where SIFT and PolyPhen predicts the variant to be benign, so the ability to use another prediction tool lowers the risk of discarding potentially important variants when establishing criteria for loss-of-function in the Predicted Deleterious filter.
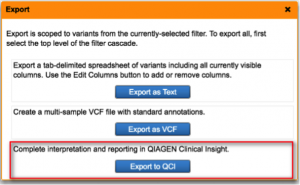
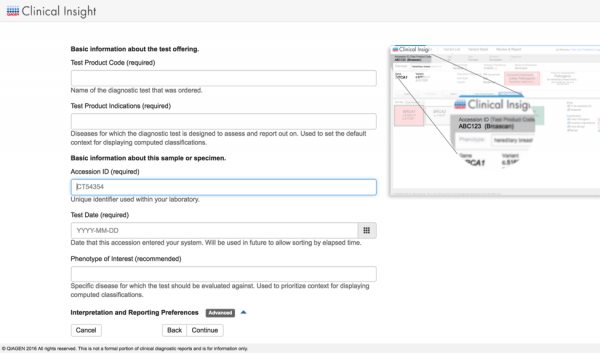
Streamline Variant Analysis to QCI Interpret workflow
QCI Interpret is a clinical decision support solution designed for genetic testing labs using next-generation sequencing platforms, catering to both somatic and hereditary cancer interpretation needs. It provides ACMG-guided variant classifications and rationale, clinical case counts from curated literature, as well as eligible treatments and clinical trials. By offering Variant Analysis users the ability to export filtered set of variants directly to QCI Interpret along with relevant metadata, we bridge the gap between variant filtering and variant reporting for clinical test offerings that involve large panels, exomes, or genomes. Prior to QCI Interpret, users can also supply data collected from additional family members or normal tissue for comparisons between case and control samples. Using the two products in unison brings efficient reduction of the candidate variant list in Variant Analysis, followed by fine-grained assessment and configurable reporting of actionable variants in QCI Interpret.
To learn more about QCI Interpret and request a demo, please visit the QCI Interpret product page.
Improvements
- Rationale used for variant inferred activity (gain/loss/normal) is presented when users click on “Inferred Activity” in the Variant Details pane
- If ExAC, AFC, or 1000 Genomes is used as part of Common Variants filtering, annotations providing homozygote counts (aggregate of all subpopulations) from these data sources are available
- Lift coverage of OMIM content from 95% to 97%, resulting in more complete references to OMIM records in Variant Findings section
- In the Biological Context filter, improved UI design for configuring inclusion of genes within 1 or more hops of the genes implicated by user-supplied terms
- In the Genetic Disease workflow’s pre-configuration for Biological Context filter, optimized order in which upstream/downstream genes are searched for compatible variants in the sample
- If a sample library is used for Controls input in an analysis set-up, the library can subsequently be used for allele frequency filtering in the Common Variants filter
- Enhanced pre-configuration settings based on best-practices guidelines for single-sample Cancer workflows
- Ability to reference existing samples in a user’s account when configuring new analyses via the API
- Access to version numbers for public content sources and QIAGEN Knowledge Base
Ingenuity Variant Analysis version 4.2.20160927
Content versions: CADD (v1.3), SIFT (2016-02-22), EVS (ESP6500SI-V2), Allele Frequency Community (2016-08-26), JASPAR (2013-11), Ingenuity Knowledge Base (Jakku 160913.000), Vista Enhancer (2012-07), Clinical Trials (Jakku 160913.000), BSIFT (2016-02-22), TCGA (2013-09-05), PolyPhen-2 (v2.2.2), 1000 Genome Frequency (phase3v5b), Clinvar (2016-06-01), COSMIC (v77), ExAC (0.3.1), HGMD (2016.2), PhyloP (2009-11), DbSNP (147), TargetScan (6.2)
Ingenuity Variant Analysis Spring 2016 Release
What’s New in the Ingenuity Variant Analysis Release (2016)
New features
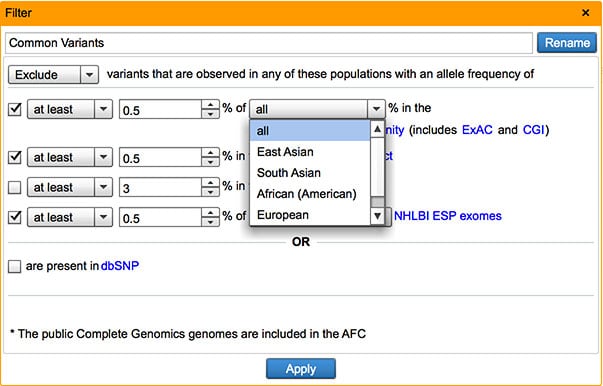
Ethnic group specific allele frequency information in Allele Frequency Community
In addition to variant composite frequency, users can now filter against East Asian, South Asian, African (American), European, and Hispanic sub-population frequencies in the Common Variants filter.
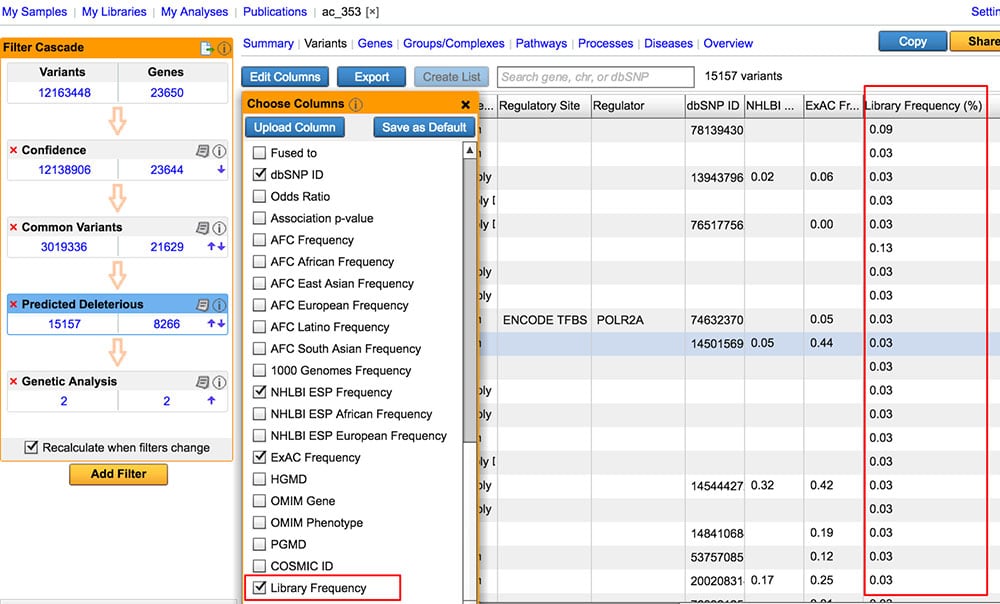
Compute allele frequencies from user-defined collection of samples
Empower users to establish their own “baseline” by computing frequencies based on a collection of samples (“My Libraries”), and annotating variants in a future analysis with library frequencies.
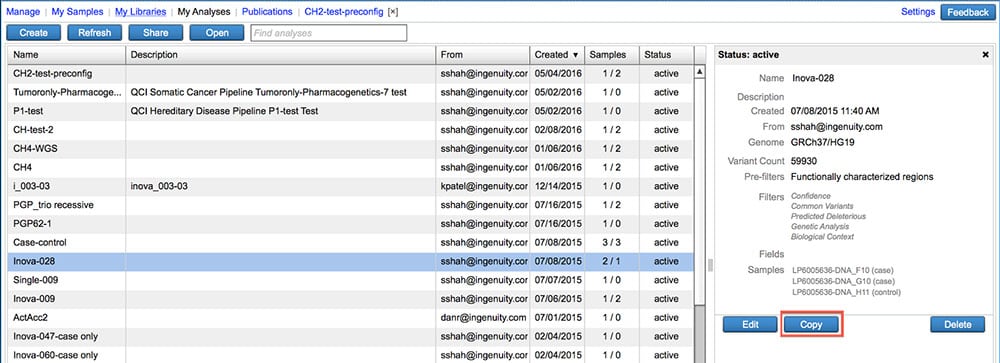
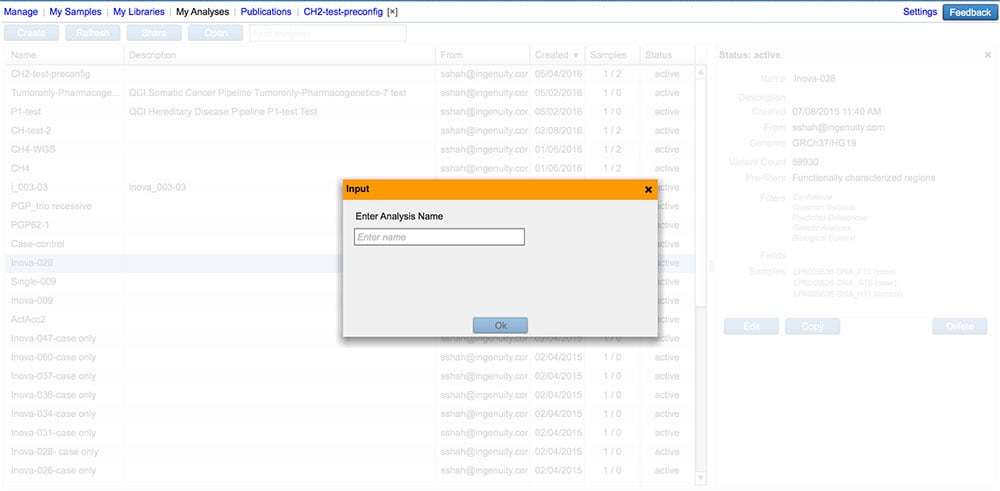
Make a copy of an existing analysis
Carry over analysis filter settings, samples, and sample metadata into a new analysis.
Improvements
- Added back the functionality to download original VCF
- Added OMIM gene and phenotype ID to the variants table
- Added GQ (Genotype Quality) to the Confidence Filter and VCF export
- Revised compound het calculation to no longer include variants of uncertain significance
- Retain allele depth (AD) information in exported VCF if uploaded VCF specifies the field
- Improved handling of analyses where prefiltering produce zero variants
- Increased numerical precision to 2 decimal points for variant call quality and allele fraction fields
- Unified syntax for Allele Frequency Community fields across application UI and exported files (now reads as “AFC frequency” or “AFC_AF”)
- API usage of Variant Analysis Custom Pipeline will now display the entire filter cascade, rather than only variants that survived the filter
Ingenuity Variant Analysis version 4.1.20160615
Content versions: CGI 54 Genomes (Version2.0), SIFT (2016-02-26), EVS (ESP6500SI-V2), Allele Frequency Community (2016-06-16), JASPAR (2013-11), Ingenuity Knowledge Base (Idris 160423.001), Vista Enhancer (2012-07), BSIFT (2016-02-26), TCGA (2013-09-05), PolyPhen-2 (v2.2.2), 1000 Genome Frequency (phase3v5b), Clinvar (2016-03-01), COSMIC (v76), ExAC (0.3), HGMD (2016.1), PhyloP (2019-11), DbSNP (146), TargetScan (6.2)